


Mystery of SARS-CoV-2 genome isolated in Bangladesh
SARS-CoV-2 has so far infected more than 4,500,000 people in 187 countries and caused over 300,000 deaths, but no drug or vaccine is yet available. In Bangladesh, over 20,000 people have been infected and 250 died. Lockdown can provide a temporary solution but we need a sustainable solution for this.
Although there are three (A,B,C) SARS-CoV-2 variants, we still don't know which one is prevailing in our country, how and through which route it has been transmitted here; if it has acquired any mutations by now and how deadly it has become. Also, we do not know why some people are affected more, showing serious symptoms, while others remain asymptomatic. We do not know why and how this has created serious havoc in some countries whereas others are only mildly affected.
In the modern era, problems in biological sciences are tackled by a bottom up approach, where we do genome sequencing of the relevant organism and associate it with other metadata to address the problem and find solutions. For the same reason so far 80 countries have deposited more than 24,000 genome sequences of this virus, which includes even countries like Nepal and Vietnam where the coronavirus problem is comparatively less severe. Since the first cases were reported on March 7, 2020 by the country's epidemiology institute IEDCR, we have been repeatedly advocating the need for genome sequencing of this virus. We also ensured that we make substantial advancement in science and technology, especially with the special attention of the prime minister in this sector.
Now we are able to do genome sequencing by Next Generation Sequencing (NGS) in our country. There are some institutes and private organisations where NGS machines are available and virus genome sequencing can be done, and also we have expert and experienced Bioinfomaticians who can perform complete genome sequence analysis. The ground-breaking work has finally been done by the Child Health Research Foundation (CHRF). Dr Senjuti Shaha and Dr Samir Kumar Shaha, along with their team from CHRF, collected samples from a 22-year-old coronavirus infected female patient and arranged to do whole genome sequencing of the virus using Illumina iSeq 100 NGS platform. As soon as the news of deposition of genome sequence data became available on May 12, Tuesday afternoon, we sought to extract this sequence and information from the public repository GISAID and CNCB, and started to explore it.
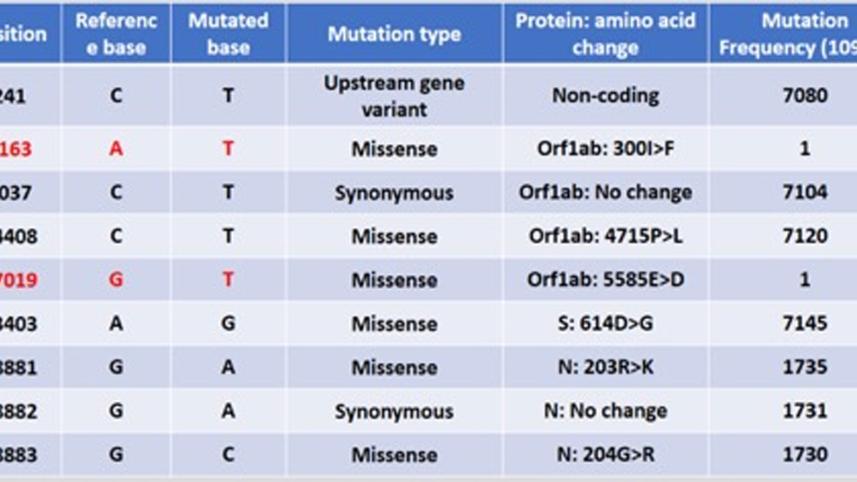
Lead by me at the Department of Genetic Engineering and Biotechnology, University of Dhaka, the Epigenetic and Bioinformatics team on nCoV research has done basic analysis of the genome. My team member Mr Abdullah Al Kamran Khan was with me in this analysis. We compared the sequence with that of the first reported coronavirus genome sequence from Wuhan, China—which is globally considered as "reference". Strikingly, we have found that this genome is very similar (99.7 percent similarity) to that of reference SARS-CoV-2 isolated from Wuhan. There are changes only in nine places and these changes are single nucleotide change (SNP). There are no deletion or insertion/addition of any large sequence compared to the original reference.
However, with great surprise, we observed that this genome has acquired two new mutations which have not been seen among the viruses reported so far and that we have observed closely. At position 1163 (genes orf1ab) a new mutation from A to T has been detected. Previously at the same position nucleotide A to C in one virus and nucleotide A to G changed in another genome reported. Also, there is a brand new mutation position at 17019 detected in our Bangladeshi isolated virus which has not been reported so far. This means that these are the new changes that the virus has acquired after entering into Bangladesh. Out of nine, seven other mutations were very common among the sequenced viruses so far. We can further study what trouble or benefit these new mutations have brought us.
Very interestingly, of these nine mutations, it contains a mutation (Single Nucleotide Mutation or SNP) in its Spike protein. There is non-silent (non-synonymous), amino acid changing (Aspartate to Glycine) mutation at the 614th position of the Spike protein (D614G). This is of particular interest because it is probably due to this mutation that the virus could spread quickly among the European and American populations compared to the original virus from China. This creates an additional serine protease (Elastase) cleavage site near the Open Reading Frame (ORF) S1 and S2 junction of the Spike protein.
The interesting aspect is that in human, a single nucleotide mutation (deletion of C nucleotide, delC) (rs35074065 variant site) in the TMPRSS2 receptor gene facilitates the entry of SARS-CoV-2 with D614G mutation to the cell very effectively. Dr Hemayet Ullah from Howard University, USA, also informed us that this delC mutation is very common in the American and European population but very rare in the East Asian/Asian populations—hence the change of amino acid aspartic acid to glycine in the S protein of the virus may be helpful for Asian countries but more infectious in the American and European populations. We do see a less severe effect in Asian countries compared to that in Europe and America. Any deleterious mutation from the perspective of an organism gets lost through natural selection and we hope more virulent mutation does not appear in Asian countries later on. Several research papers are also available on this mutation.
To understand the origin, we have constructed phylogenetic tree (UPGMA and Neighbour-Joining) in MEGA with default parameters, with representative sequences from 60 other countries and the reference sequence, totalling 350 sequences. Phylogenetic tree shows that this Bangladeshi SARS-CoV-2 genome isolate seems closer to European cluster—most likely the person got infected from someone who returned from Europe or maybe she herself returned from there. We are fine-tuning the phylogenetic tree. And are also in the process of making phylogenetic tree with 10,000 high quality sequences selected from 80 countries to better explain the origin and route of transmission of this particular virus.
To understand the pattern of infection in Bangladesh, only one genome sequence is not enough. We need sequence of at least 100 isolates. We have made a proposal to the ICT ministry in response to their "Call for Nation (Hakathon)". In this study proposal we aim to create a dataset by combining 100 coronavirus genomes from Bangladeshi patients and integrate this genome information with patient's personal/clinical/treatment/diagnostic and other information. This information will be analysed extensively by computational methods to do clustering, phylogenetic and pharmacogenomics studies, and will compare data with other globally available data to make a concrete information-base that will help pharmaceutical industries produce appropriate drugs and vaccines for our population.
Also, the ICT ministry will be able to announce that Bangladesh has uncovered the genome mystery of the coronavirus circulating in the country and trace back the transmission. This project will be a multicentre research where essential help from ICT/Bangladesh government, and help of IEDCR through the government will be required to get patients' samples and relevant clinical data. We will carry out sequencing (Next Generation Sequencing) of the viral genome and other analyses with our own resources in Bangladesh. If ICT/government support us, it is also possible to do further research in future where in addition to the viral genome we can sequence genome of some individuals who were infected and developed the disease as well as healthy individuals who did not develop the disease. This may also let us know the factors (if any) that conferred resistance to them.
Dr ABMM Khademul Islam, associate professor, Genetic Engineering and Biotechnology, University of Dhaka.
 For all latest news, follow The Daily Star's Google News channel.
For all latest news, follow The Daily Star's Google News channel.
Comments